using XAM
using BioAlignments
using GenomicFeatures
ch1df=DataFrame(refname=String[],position=Int64[],rightposition=Int64[],sequence=String[])
bamfile="/share/home/zhangchunyong/proj/Zhulab/epimutation/data/test.bs.sortedbychr.bam"
reader=open(BAM.Reader,bamfile)
for record in reader
# `record` is a BAM.Record object.
if BAM.ismapped(record)
a=BAM.refname(record), BAM.position(record),BAM.rightposition(record),BAM.sequence(record)
push!(ch1df,a)
end
end
我的目的是从一个bam文件中筛选出那几列,有没有更高效的方法,总感觉这样一行一行push很慢
这种用法我没试过,但是根据我的经验吧,每次push!的时候,DataFrame都会改变它的大小——进而每次都要重新分配内存。
你试试一开始就把大小、数值类型固定住。这样他内存就固定了。
然后你再逐表格去赋值试试。
应该快很多。
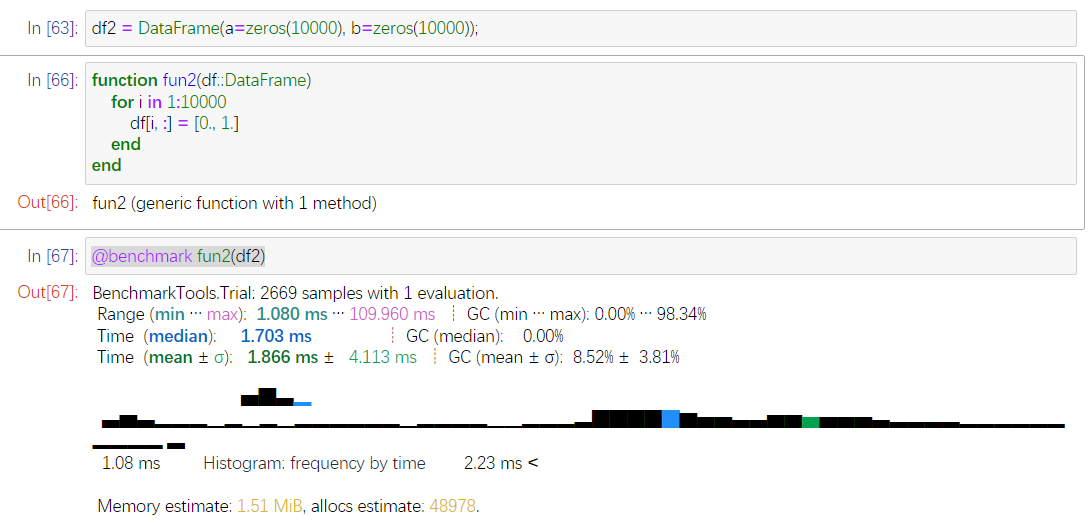
你好,我测试了一下,结论是:更改Vector元素值比更改DataFrame更快,建议暂时别直接用DataFrame。
同样10000×2的数据,
通过10000次push!
循环的平均
4.571ms。可以看到用来分配内存就有80000次。
通过预分配,在更改10000次DataFrame值
循环平均要
1.866ms。但是用来分配内存的还有48978次。
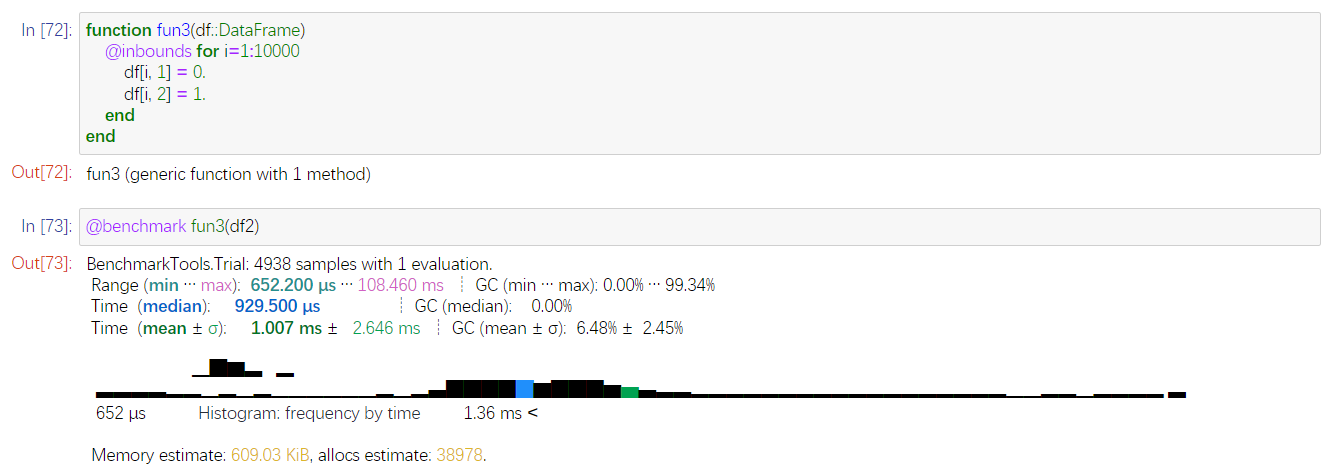
我想到元组打包的时候,也是要分配内存的。比如你的
a=BAM.refname(record), BAM.position(record),BAM.rightposition(record),BAM.sequence(record)
就会把右边4个打包,打包时候也会分配内存。
这里是一个元素一个元素去改,省去打包过程,发现平均需要
1ms。但是内存还有38978次分配。
不知道哪儿来的,有大佬可以解答一下吗?
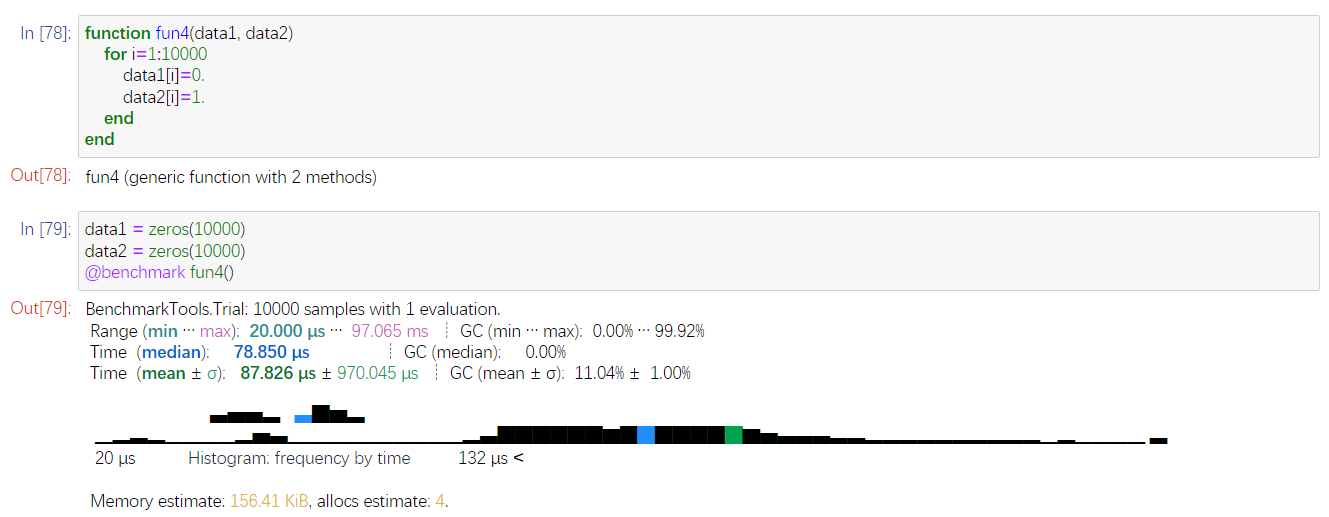
最推荐后面这个了。先保存到Vector中,在Vector中改值,再将Vector改成DataFrame。
测试下来,只要
87.826μs,快了50多倍。
当然这个只是我对保存数据的部分提出的建议。但是我其实感觉你读取数据部分也很费时间,所以实际速度应该比这个低。
有点看法:
- 这里对
record这个变量需要调用4次函数,就不能一个函数同时得到4个吗?
- 我不知道每次通过
.来访问会不会降低速度。有大佬懂的可以解答下。
- 建议读读官方文档的性能建议部分。里面有一点 分离核心函数(又称为函数屏障)。就是鼓励你将最耗时间的部分写成函数,
Julia会针对这个进行优化。
- 试试并行。在
for循环外面,根据reader的行数,将Vector的内存分配好——好多线程大家一起push!可能会出问题——然后用Threads.@threads装饰for循环。然后对index进行循环读取数据,和更改Vector的值。最后保存成DataFrame格式。然后关掉这个文档,用julia -t 10 XXX.jl启动这个脚本。
其实多线程并行我没用过,这里分享我记的笔记:
多线程启动终端
julia -t 10
多线程启动脚本
julia -t 10 test.jl
多线程装饰器
Threads.@threads # 装饰for循环
1 个赞